
Reliable pKₐ Prediction through Efficient Incorporation of Anharmonicity within the Nuclear–Electronic Orbital Framework
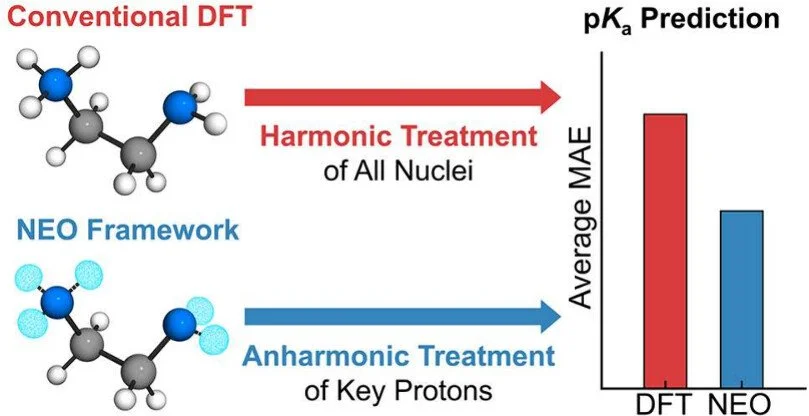
Accurate pKₐ prediction is critical for understanding chemical reactivity and molecular properties across a wide range of applications. Computational approaches usually invoke a harmonic treatment of the vibrational modes for zero-point energies, as well as thermal and entropic contributions. Herein, we present a general protocol for relative pKₐ prediction that incorporates the significant anharmonic effects using nuclear–electronic orbital (NEO) theory. This protocol is validated against experimental data for a range of molecules in acetonitrile, including protonated nitrogen bases, nitrophenols, anilines, and diamines, as well as cobalt electrocatalysts. For simple acids, the NEO approach offers only a slight improvement over conventional density functional theory with the standard harmonic vibrational treatment, whereas for hydrogen-bonded acids, the NEO approach offers more significantly improved performance at a comparable computational cost. This accessible methodology provides a practical route for accurate pKa prediction in challenging systems and is extendable to related thermodynamic properties such as hydricities and proton-coupled redox potentials.
You, J. M.; Chow, M.; Paenurk, E.; Hammes-Schiffer, S. Reliable pKa Prediction through Efficient Incorporation of Anharmonicity within the Nuclear–Electronic Orbital Framework, J. Am. Chem. Soc., 2025, 147 (40), 36059-36065. https://doi.org/10.1021/jacs.5c11332