
Constrained nuclear–electronic orbital method for periodic density functional theory: Application to H₂ chemisorption on Si(001) surfaces
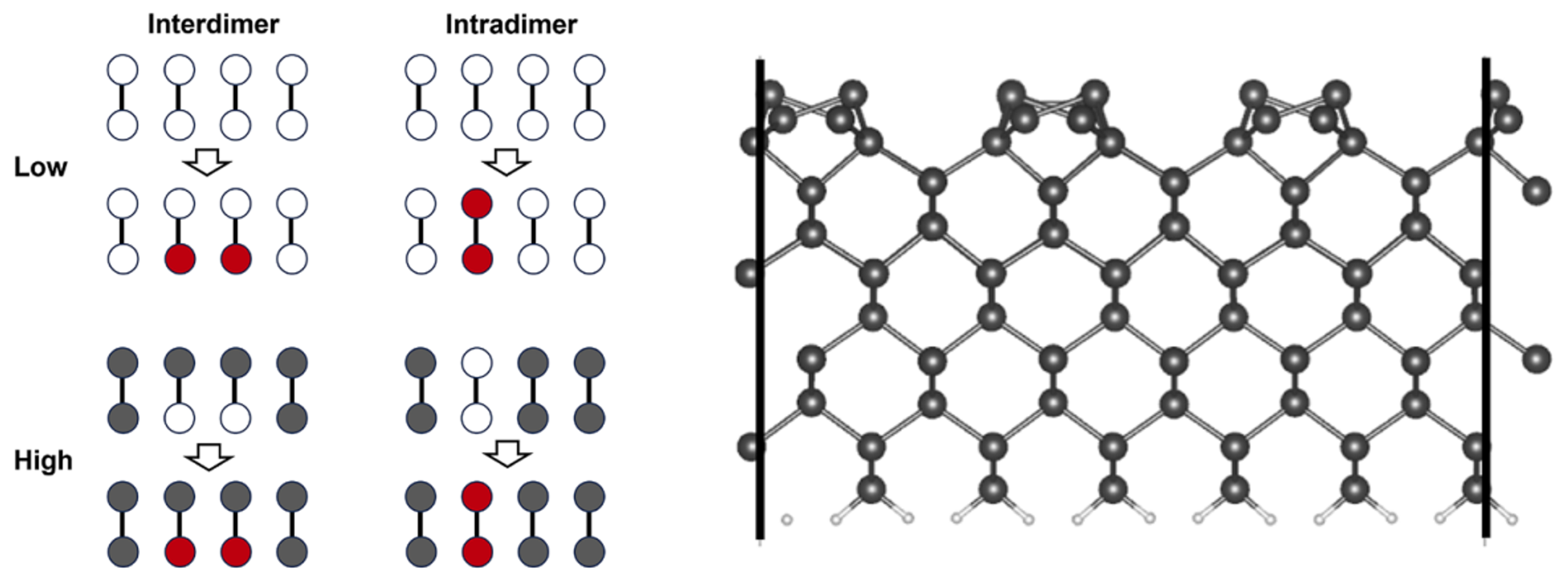
The nuclear–electronic orbital (NEO) method provides a powerful computational framework for incorporating nuclear quantum effects (NQE) in electronic structure calculations beyond the Born–Oppenheimer approximation. By incorporating additional constraints to the position operator on quantum particles like protons, the NEO method enables calculation of effective potential that accounts for NQE. In this work, we present a new constrained NEO (cNEO) formulation for density functional theory (cNEO-DFT) calculations in the context of extended periodic systems. Using the nudged elastic band method, we discuss an application of the cNEO-DFT approach to studying the adsorption of a hydrogen molecule on the Si(001) surfaces. The calculation shows how NQE impacts the reaction energetics. The proton density changes are computed along the reaction pathways. This work demonstrates the capability of the new cNEO-DFT method to study a wide range of chemical processes, such as surface reactions where the quantum nature of light atoms like protons is non-negligible.
Liu, S.; Xu, J.; Kanai, Y. Constrained Nuclear-Electronic Orbital Method for Periodic Density Functional Theory: Application to H2 Chemisorption on Si(001) Surfaces, J. Chem. Phys., 2025, 163, 084110. https://doi.org/10.1063/5.0278375